What is alkaptonuria?
|
Alkaptonuria is a genetic disorder that affects the metabolism of homogentisic acid, causing it to build up in the body. Homogentisic acid is an intermediate in the pathway for breaking down phenylalanine and tyrosine, both of which are found in many dietary proteins. This buildup causes damage to cartilage throughout the body, can induce the formation of kidney stones, and cause urine to turn black after being exposed to oxygen [1].
Those with alkaptonuria are often asymptomatic until adulthood, however pigmentation in the sclera of the eye and cartilage of the ear may be noted. Serious symptoms often begin with pain in the knees, back, and hips as cartilage is damaged by the homogentisic acid. Calcification of the heart valves, irregularities in heart rhythm, and kidney stones are other common symptoms. These symptoms greatly affect quality of life for affected individuals, with many needing joint replacement surgery before age 60 [1]. |
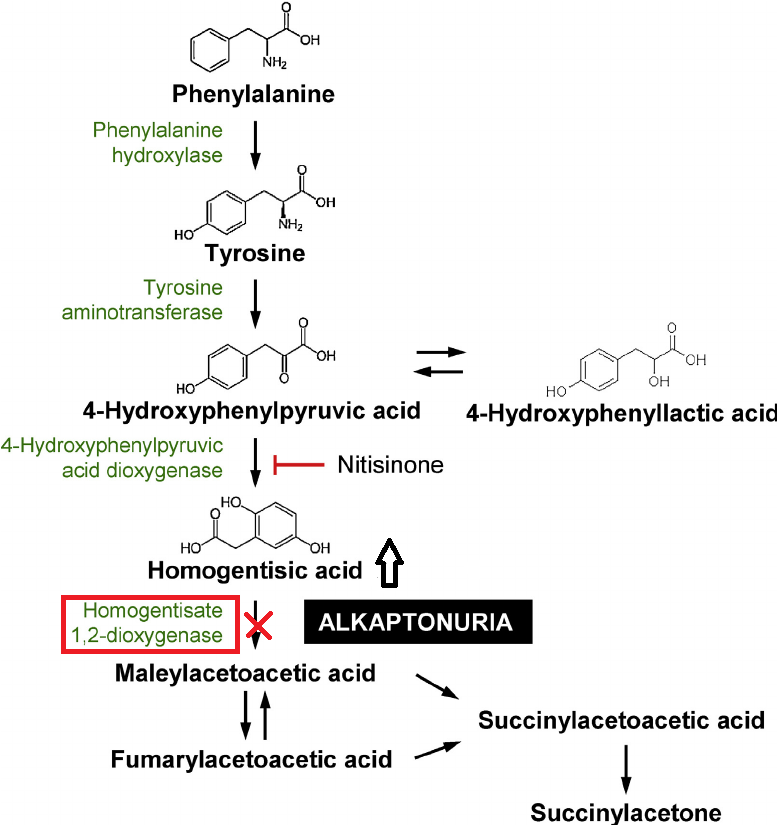
Figure 1. The metabolic breakdown pathway of phenylalanine and tyrosine is shown above, including a depiction of where in the pathway the alkaptonuria-causing disruption occurs and the 3-hyroxyphenylpyruvic acid dioxygenase inhibition effects of Nitisinone.
|
What causes alkaptonuria?
A mutation in the gene HGD, which encodes the homogentisate 1,2-dioxygenase enzyme, is the cause for alkaptonuria. It is an autosomal recessive disorder, meaning an individual must have two copies of the mutated gene in order to be affected by the disease. Alkaptonuria affects around 1 in every 250,000 people, but the rates of incidence are much higher in the Dominican Republic and Slovakia [2].
HGD is found on the long arm of chromosome 3. There are over 65 mutations in HGD that are associated with alkaptonuria, with the most common among Europeans being a substitution of methionine with valine at protein position 368 [3]. The genetic basis for the disease was not discovered until 1996 [2].
HGD is found on the long arm of chromosome 3. There are over 65 mutations in HGD that are associated with alkaptonuria, with the most common among Europeans being a substitution of methionine with valine at protein position 368 [3]. The genetic basis for the disease was not discovered until 1996 [2].
Diagnosis and treatment
|
Diagnosis of alkaptonuria can occur by taking urine samples and analyzing the amount of homogentisic acid found. As mentioned above, pigmentation and pain in the joints can also be used as indicators of the disease's presence. A questionnaire called the AKU Severity Score Index can be used to quantify the severity of symptoms and the patient's response to treatment. All treatments options for the disease treat only the symptoms and not the underlying condition. Even restricting the amount of dietary protein, a difficult task, has been shown to be an ineffective treatment [1].
The drug Nitrisinone has been suggested as a treatment option, as it inhibits the enzyme used to produce homogentisic acid, but continued use would lead to a buildup of tyrosine (the effects of which are currently unknown) [1]. |
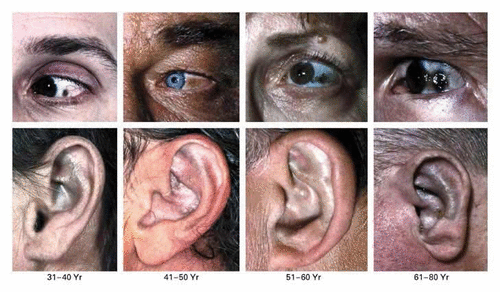
Figure 2. The effects of alkaptonuria on skin and eye pigmentation at different ages. Symptoms such as joint pain typically onset at around age 40.
|
|
William Rosenthal
University of Wisconsin-Madison, 2018 [email protected] Page updated: 4-5-2018 http://www.genetics564.weebly.com |
References:
[1] Ranganath, L. R., Jarvis, J. C., & Gallagher, J. A. (2013). Recent advances in management of alkaptonuria (invited review; best practice article). Journal of Clinical Pathology, 66(5), 367-373. doi:10.1136/jclinpath-2012-200877 [2] Zatkova, A. (2011). An update on molecular genetics of Alkaptonuria (AKU). Journal of Inherited Metabolic Disease, 34(6), 1127-1136. doi:10.1007/s10545-011-9363-z [3] HGD gene - Genetics Home Reference. (n.d.). Retrieved February 08, 2018, from https://ghr.nlm.nih.gov/gene/HGD#resources |
Image references:
Figure 1: https://steemit-production-imageproxy-upload.s3.amazonaws.com/DQmf4WVnp8buHeKFQtddq9gi6BcyxYK5MiFa1ADgfpfznPA Figure 2: http://www.nejm.org/na101/home/literatum/publisher/mms/journals/content/nejm/2002/nejm_2002.347.issue-26/nejmoa021736/production/images/medium/nejmoa021736_f3.gif |
Home About DONATE

